|
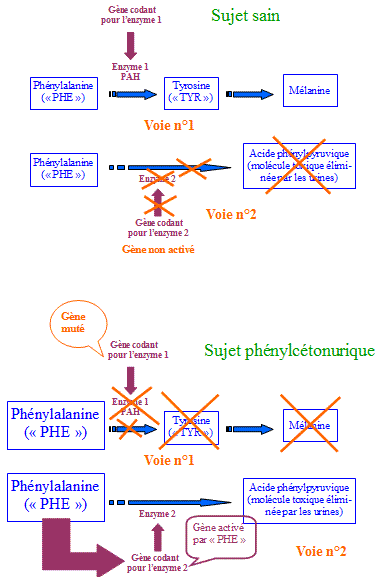
La phénylcétonurie est une affection héréditaire causée par la perturbation du métabolisme d'un acide aminé, la phénylalanine ("PHE"), que contiennent certains aliments. Elle atteint 1 nouveau-né sur 10 000 et se manifeste dès la première semaine par des troubles digestifs, des lésions cutanées et des convulsions. Elle provoque un important retard mental et l'espérance de vie sans traitement ne dépasse pas vingt ans. Les sujets atteints de phénylcétonurie sont caractérisés par la pâleur de leur visage, leurs yeux et leurs cheveux très clairs, même dans les populations où ces caractères sont totalement inhabituels. Ces sujets malades ont un taux anormalement élevé de phénylalanine et dacide phénylpyruvique dans le sang et dans lurine.
Deux voies de dégradation métabolique de la phénylalanine dans l'organisme sont connues : la voie n°1 chez les sujets sains (l'enzyme 1 (= phénylalanine hydroxylase ou PAH) est décelable dans les cellules hépatiques (= du foie) de l'individu sain) et la voie métabolique n°2 qui n'est utilisée qu'en cas d'augmentation anormale du taux de phénylalanine dans le sang.
|
|
 |
(fermer la fenêtre) |